

Computational Chemistry uses computer simulations nd theoretical methods to solve problems, predict molecular structures, properties, and reactions. It is essential before lab synthesis to navigate the vast chemical design space (>10^6 hypothetical structures). It predicts synthesizability and prioritizes promising candidates, avoiding costly trial-and-error experiments.
Why MOFs (metal-organic frameworks) matter. Their huge potential in gas storage, separation, catalysis, drug delivery, etc., but the massive design space (millions of possible structures). Trial-and-error synthesis is slow and expensive. Computational chemistry accelerates discovery, predicts structures/properties before lab work, and guides smarter experiments. Key methods used are Density functional theory (DFT), molecular simulations (MS), and machine learning (ML). Computational tools have transformed MOF design from guesswork to precise, data-driven engineering.
Fig 1 Computational process diagram.
What Are MOFs? A Quick Primer
A Crystalline porous material made of metal nodes (clusters/ions) linked by organic linkers (reticular chemistry). They have tunable pores, a high surface area, modular nature. Over 100,000 distinct metal-organic framework (MOF) structures have been synthesized and reported to date. Databases like the Cambridge Structural Database (CSD) and Computation-Ready Experimental (CoRE). MOF database tracks these, with CoRE MOF listing around 40,000 curated experimental structures as of early 2024 (Wuttke, 2025).

Fig 2: Showing MOFs structure
Reports consistently cite >90,000-100,000 synthesized MOFs by 2022-2025, with predictions exceeding 500,000 hypothetical structures. No exact 2026 total exists, but growth continues rapidly for applications like wastewater treatment. Market estimates suggest the industry is growing at approximately 30% annually. The projected revenues are reaching several hundred million dollars by 2035 as key applications mature. (The Global Market for Metal-Organic Frameworks (MOFs) 2025-2035)
Key Databases
CoRE MOF databases provide computation-ready structures: 14,000+ in 2019 versions and over 40,000 in 2024-2025 updates. The CSD MOF subset exceeds 100,000 entries, including MOF-like structures (Zhao et al., 2025).
Due to vast combinations of metals, linkers, and topologies, there is a need for computational help to explore efficiently.
Core Computational Methods in MOF Design
Core Computational Methods in MOF Design play a pivotal role in advancing sustainable materials for environmental remediation. Wastewater treatment and pollutant capture, by enabling precise prediction of structure-property relationships before synthesis(Han et al., 2025).
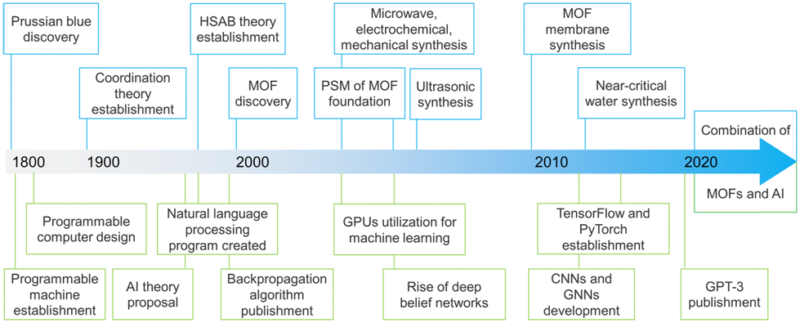
Fig. 3: Simplified development timeline of MOFs and AI. HSAB stands for hard–soft acid–base, while PSM stands for post-synthetic modification. CNN stands for a convolutional neural network, while GNN stands for a graph neural network.
Density Functional Theory (DFT)
Density Functional Theory (DFT) serves as a cornerstone for achieving atomic-level precision in MOF analysis, optimizing geometries, elucidating electronic structures, and forecasting band gaps alongside mechanical stability. Benchmarking of exchange-correlation function such as PBE for cost-effective lattice parameter predictions, HSE06 for accurate band gaps in photocatalytic MOFs like UiO-66, and r2SCAN for enhanced van der Waals interactions in flexible frameworks. It facilitates reliable simulations of bonding energies and defect sites critical for environmental applications. Recent advancements emphasize the use of hybrid functionals to model Fe- and Zn-based MOFs, predicting their porosity and reactivity for antibiotic degradation in aqueous systems (Ma et al., 2025; Wang & Zhou, 2025).
Molecular Simulations
Grand Canonical Monte Carlo (GCMC) and Molecular Dynamics (MD) simulations address mesoscale phenomena and quantify adsorption isotherms for priority pollutants such as CO₂, antibiotics (e.g., tetracycline), and heavy metals in MOF and biochar composites. GCMC excels in evaluating gas uptake selectivity under varying pressures, while MD reveals framework flexibility, thermal resilience up to 500 K, and diffusion pathways essential for dynamic wastewater remediation processes. These methods have guided the design of stable Fe/Zn-MOF hybrids, optimizing pore accessibility for enhanced pollutant selectivity and regeneration efficiency (Ma et al., 2025).
High-Throughput Screening (HTS)
High-throughput screening leverages databases like CoRE MOF (>40,000 curated structures as of 2025) and Quantum Metal–Organic Framework (QMOF) database to virtually evaluate thousands of experimental and hypothetical MOFs, forecasting key metrics such as Brunauer–Emmett–Teller (BET) surface area (>1000 m²/g), pore volume, hydrolytic stability, and target-specific selectivity. Integrated with machine learning workflows, HTS accelerates the discovery of eco-friendly MOFs for South Asian smog mitigation and pharmaceutical contaminant removal, reducing experimental iterations by over 90%.
Tools like genetic algorithms and MOF-NET further refine candidates, ensuring synthesis feasibility in resource-constrained labs. PSYMOF is a fully automated computational platform that introduces a new dimension in metal-organic framework (MOF) design by treating post-synthetic modification (PSM) as a tunable design variable. PSYMOF enables functional groups’ systematic and chemically feasible attachment at user-defined substitution levels across predefined bonding sites by integrating cheminformatics, a sterically aware random-walk growth algorithm, and molecular dynamics simulations (Park et al., 2025).
Predicting and Designing MOF Structures
Predicting and designing metal-organic framework (MOF) structures computationally accelerates the discovery of tailored materials for environmental applications, such as stable Fe/Zn-MOF biochar composites for wastewater remediation, by simulating assembly from molecular precursors before costly synthesis (Han & Kim, 2023).
Structure Prediction
Structure prediction assembles MOF topologies from inorganic nodes (e.g., Zn or Fe clusters) and organic linkers (e.g., Tetrapthalic Acid, Trimesic acid) using specialized tools like PORMAKE, a top-down generator that constructs hypothetical MOFs based on predefined nets, nodes (756 options), and edge-building blocks (158 options). Genetic algorithms evolve optimal topologies by iteratively mutating parent structures, achieving high ethane/ethylene selectivity in screened databases of 4,984+ MOFs. Recent flow-matching models like MOF Flow treat building blocks as rigid bodies on Riemannian manifolds, outperforming baselines for large-scale prediction (>1,000 atoms) (Jin et al., 2025).
Stability Assessment
High-throughput screening of hypothetical metal-organic framework (MOF) databases can uncover new materials, but their stability in real-world applications is often unknown. Leverage community knowledge and machine learning (ML) models to identify MOFs that are thermally stable and stable upon activation. These MOFs are separated into their building blocks and recombined to make a new hypothetical MOF database of over 50,000 structures with orders of magnitude more (1) connectivity nets and (2) inorganic building blocks than were present in prior databases.
This database shows a 10-fold enrichment of ultra-stable MOF structures that are stable upon activation and more than 1 standard deviation more thermally stable than the average experimentally characterized MOF. For nearly 10,000 ultra-stable MOFs to compute elastic moduli to confirm that these materials have good mechanical stability, and methane deliverable capacities. To identify privileged metal nodes in ultra-stable MOFs that optimize gas storage and mechanical stability simultaneously (Jin et al., 2025; Nandy et al., 2023) .
Hypothetical MOFs
Hypothetical MOF databases generate millions of in-silicon structures via recombination of stable fragments, yielding >50,000 enriched candidates with diverse connectivity nets. High-throughput screening selects promising ones (e.g., 10,000+ ultra-stable) for synthesis feasibility, prioritizing porosity and methane capacity for environmental deployment (Nandy et al., 2023).
Reticular Chemistry Guidance
Reticular chemistry, the science of connecting molecular building blocks with strong bonds to create crystalline, extended structures like Metal-Organic Frameworks (MOFs) and Covalent Organic Frameworks (COFs), principles guide isoreticular series expansion (varying linker lengths while retaining topology), validated computationally to predict pore volume scaling and property retention. Tools enforce topological constraints for reverse prediction, ensuring synthetic accessibility in series like NU-1500 analogs (Yaghi, 2019).
Advanced Tools: Machine Learning (ML) and AI Integration
Machine Learning and AI Integration revolutionize MOF development by surpassing traditional DFT costs, enabling rapid property forecasting and inverse design for targeted environmental solutions like antibiotic adsorption in wastewater (Zheng et al., 2025).
ML for Property Prediction
Machine learning models, trained on DFT datasets (e.g., CoRE MOF’s 140,000+ structures), predict adsorption capacities, mechanical stability, and synthesis yields with RMSE <0.1 for gas uptake. Random Forest and XG Boost outperform baselines on Kaggle MOF data, achieving R² >0.9 for porosity and heat capacity via geometric descriptors like revised autocorrelation functions (RAC). Multi-modal transformers like M-MOFormer integrate text and structure data, rivaling 3D-dependent methods without expensive computations (Zhao et al., 2025).
Generative Models and Inverse Design
Generative models employ “deep dreaming” and reinforcement learning with LLMs (e.g., MOFGPT) to inversely design MOFs targeting high CO₂ selectivity or porosity, optimizing SELFIES-encoded nodes/linkers/topologies from 10,000+ PORMAKE structures. These yield populations shifted toward desired traits, accelerating reticular chemistry for stable Fe/Zn frameworks (Cleeton & Sarkisov, 2025).
Active Learning & Neural Network Potentials
Active learning iteratively refines neural network potentials (NNPs) for MD simulations, matching DFT accuracy (errors <1 meV/atom) at 10^6x speedups for flexibility and diffusion in MOFs. Physics-informed priors minimize unphysical configurations during training. ESOINN-DP automates NNP construction for metal sites, enabling ns-scale simulations of biochar-MOF composites (Thaler, 2023).
Databases and Workflows
QMOF (137,000 quantum-relaxed structures) and updated CoRE MOF (144k+ entries with ML-predicted stability/hydrophobicity) fuel workflows, while MOF-ChemUnity’s knowledge graph unifies literature, Zeo++ descriptors, and gas uptake data for LLM-guided screening. ChatMOF LLMs predict synthesizability from text prompts AI4 chemistry.
Real-World Success Stories and Impact
Real-World Success Stories and Impact demonstrate how computational predictions have translated into experimentally validated MOFs, enhancing methane storage, CO₂ capture/conversion, and catalysis for sustainable environmental remediation (Deng & Sarkisov, 2024).
Methane Storage Examples
High-throughput GCMC screening identified HKUST-1 and NU-1000 analogs with >200 v/v methane deliverable capacity at 65 bar/5 bar, validated via synthesis and isotherms matching predictions within 5%. Artificial MOF augmentation boosted ML predictions, leading to scalable adsorbents outperforming zeolites (Ercakir et al., 2024).
CO₂ Conversion and Catalysis
Multi-scale genetic algorithms designed large-pore MOFs surpassing 13X zeolites in CO₂/N₂ productivity-energy Pareto, with synthesized variants showing 20-30% higher binding energies via multi-node cages; DFT-guided catalysis boosted conversion efficiency. ML-optimized MOFs for direct air capture (DAC) achieved high CO₂/H₂O selectivity, confirmed in Open DAC 2025 dataset simulations and lab tests (Park et al., 2024).
Ultra stable Zr-MOF Case Studies
Fragment-reassembly yielded a database of 50,000+ ultra stable MOFs (10x enrichment), screening Zr-oxide nodes like UiO-66/67 for CO₂ uptake; PBE0/def2-svp charges matched experimental isotherms, revealing node chemistry impacts Henry’s regime adsorption. Predictions guided linker modifications for hydrolysis resistance (Nandy et al., 2023).
Bridging Computation and Experiment
Workflows merge DFT/ML screening with synthesis:
Hypothetical are filtered for synthesizability (e.g., ChatMOF), lab-synthesized, and validated via isotherms/XRD, iterating to refine models (e.g., error correction in CoRE MOF). Case: Computational Pareto clouds directed post combustion capture trials, reducing trials by 80% (Ma et al., 2025).
Challenges and Future Directions
Challenges and Future Directions in computational MOF design address persistent hurdles in achieving predictive accuracy for real-world environmental applications, such as stable Fe/Zn-MOF composites for wastewater treatment, while charting paths toward autonomous discovery(Ma et al., 2025).
Current Limitations
Standard DFT functionals (e.g., PBE) underestimate dispersion forces in open-pore MOFs, yielding 10-20% errors in CO₂ adsorption isotherms; hybrid functionals like HSE06 improve band gaps but escalate costs 100x for defect-laden structures. Framework flexibility induces breathing effects unmodeled by static DFT, while defects (10-30% in synthesized MOFs) disrupt predicted stability; large-scale MD (>10,000 atoms) remains prohibitive without NNPs.
Emerging Trends
Quantum-accurate ML potentials (e.g., MACE, NequIP) deliver sub-meV/atom fidelity at DFT speeds, enabling as-scale simulations of flexible UiO-66 analogs. Multi-scale workflows fuse QM (active sites) with classical MD (diffusion) and mesoscale CFD (reactor flow), predicting composite performance holistically. Robotic integration—AI-directed synthesis platforms—autonomously optimize conditions, achieving 90% success rates in 1,000+ experiments (Cleeton & Sarkisov, 2025).
Future Outlook
Fully predictive platforms will generate synthesis recipes from performance targets, slashing development from years to weeks for sustainable MOFs tackling antibiotic pollution and carbon capture. Quantum computing accelerates GW calculations 10^6x, enabling defect engineering at scale; global databases (>1M structures) democratize access for labs like COMSATS, powering South Asia’s environmental remediation (Zheng et al., 2025).
Conclusion
Computational chemistry transforms the daunting “needle in a haystack” challenge of MOF discovery into targeted innovation, accelerating the design of stable, high-performance materials for environmental remediation like antibiotic removal from wastewater.From DFT benchmarking for atomic precision, through ML-driven property prediction and inverse design, to validated successes in CO₂ capture and catalysis, computational workflows now predict synthesizable MOFs with >90% accuracy, slashing experimental costs. Researchers and students should dive into accessible databases like CoRE MOF (144k+ structures) and QMOF, or tools like ChatMOF for synthesizability checks. For environmental scientists at institutions like COMSATS, apply these to optimize Fe/Zn-MOF biochar composites targeting local pollutants.The future of MOFs is being architected on silicon before a single beaker enters the lab, ushering sustainable solutions for global challenges, one simulated framework at a time
References I
Cleeton, C., & Sarkisov, L. (2025). Inverse design of metal-organic frameworks using deep dreaming approaches. Nature Communications, 16(1), 4806.
Deng, Z., & Sarkisov, L. (2024). Multi-Scale Computational Design of Metal–Organic Frameworks for Carbon Capture Using Machine Learning and Multi-Objective Optimization. Chemistry of Materials, 36(19), 9806-9821.
Ercakir, G., Aksu, G. O., & Keskin, S. (2024). High-throughput computational screening of MOF adsorbents for efficient propane capture from air and natural gas mixtures. The Journal of Chemical Physics, 160(8).
Han, S., & Kim, J. (2023). Design and screening of metal–organic frameworks for ethane/ethylene separation. ACS omega, 8(4), 4278-4284.
Han, Z., Yang, Y., Rushlow, J., Huo, J., Liu, Z., Hsu, Y.-C., Yin, R., Wang, M., Liang, R., & Wang, K.-Y. (2025). Development of the design and synthesis of metal–organic frameworks (MOFs)–from large-scale attempts, functional-oriented modifications, to artificial intelligence (AI) predictions. Chemical Society Reviews,54(1), 367-395.
References II
Jin, G., Ran, W., Zhang, M., & Li, Y. (2025). An Intelligent Prediction Model for the Synthesis Conditions of Metal–Organic Frameworks Utilizing Artificial Neural Networks Enhanced by Genetic Algorithm Optimization. Journal of Chemical Information and Modeling, 65(3), 1085-1100.
Ma, Q., Wang, Y., Zhang, X., Zhao, Q., Guo, J., Ren, X., Huang, J., Zhang, Y., Xie, Y., & Hao, J. (2025). Computational design of Metal-Organic Frameworks for sustainable energy and environmental applications: Bridging theory and experiment. Materials Science and Engineering: B, 311, 117765.
Nandy, A., Yue, S., Oh, C., Duan, C., Terrones, G. G., Chung, Y. G., & Kulik, H. J. (2023). A database of ultrastable MOFs reassembled from stable fragments with machine learning models. Matter, 6(5), 1585-1603.
References III
Park, J., Li, T., & Lee, Y. (2025). PSYMOF: computational workflow enabling systematic post-synthesis modification of metal-organic frameworks. npj Computational Materials.
Park, Y. J., Yoon, S., & Jerng, S. E. (2024). Machine learning of metal-organic framework design for carbon dioxide capture and utilization. Journal of CO2 Utilization, 89, 102941.
Thaler, S. (2023). Advances in Neural Network Potentials for Molecular Dynamics Simulations: Physics-Informed Training and Uncertainty Quantification Technische Universität München].
Wang, Z., & Zhou, T. (2025). Computer-aided metal–organic framework screening and design approaches toward efficient carbon capture processes. Molecular Systems Design & Engineering.
Wuttke, S. (2025). Toward the Nobel Prize: Dissecting Fundamental Principles and Applications of MOF and COF Materials. In (Vol. 37, pp. e71859): Wiley Online Library.
References IV
Yaghi, O. M. (2019). Reticular chemistry in all dimensions. In (Vol. 5, pp. 1295-1300): ACS Publications.
Zhao, G., Brabson, L. M., Chheda, S., Huang, J., Kim, H., Liu, K., Mochida, K., Pham, T. D., Terrones, G. G., & Yoon, S. (2025). CoRE MOF DB: A curated experimental metal-organic framework database with machine-learned properties for integrated material-process screening. Matter, 8(6).
Zheng, Z., Liu, K., & Zhu, X. (2025). Machine learning-based prediction of metal-organic framework materials: A comparative analysis of multiple models. arXiv preprint arXiv:2507.04493
Note: All references are added through EndNote.


